『 医療機器ソフトウェアのリファクタリング』という動画を作った。
この動画を作成するにあたっては、第8回 組込みシステム開発技術展(ESEC) 2005年6月29日~7月1日 専門セミナー で『高信頼性を確保するソフトウェア開発手法と実践』という講演をしたときの スライドを使った。
20年前に作ったスライドだが、このときのスライドが今でも使えるということは、ソフトウェア開発の世界はそんなに進化していないのかもしれない。
この動画を作ったきっかけは、医療機器ソフトウェア(SaMD)を新規に開発しようとするスタートアップ企業は、まず、フィージビリティスタディとしてソフトウェアを作ってしまい、いざ、薬機法申請するために 規制要件である IEC 62304(医療機器ソフトウェアーソフトウェアライフサイクルプロセス)に適合しようとするという話しを聞いたからである。

IEC 62304 は、リスクベースアプローチなので、そのものが不具合を起こしたときに患者危害に至るようなソフトウェアモジュールはリスクレベルが高い(ソフトウェア安全クラスC)とし、もっとも厳しいアクティビティ、タスクを課す。

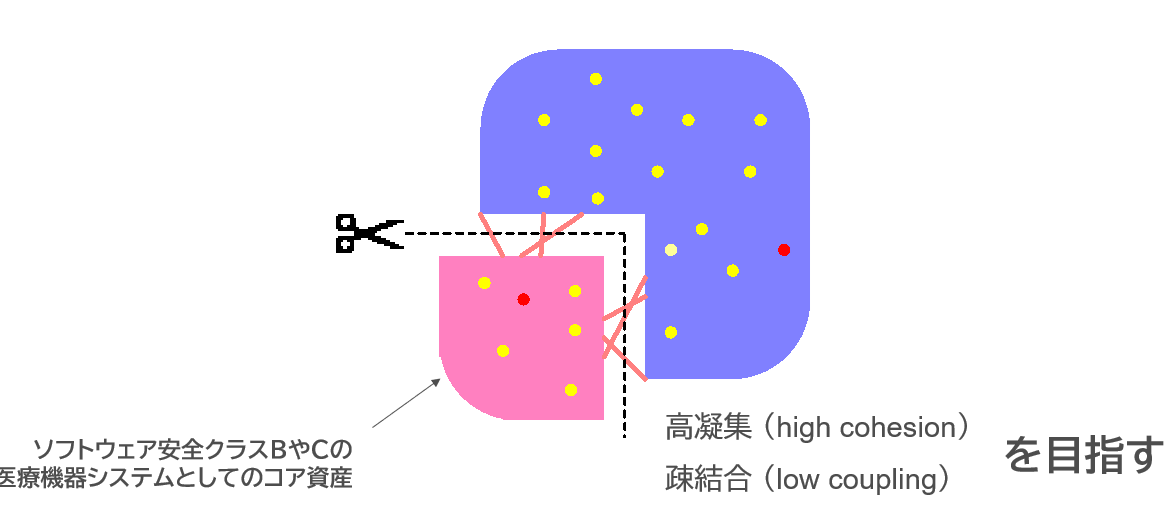
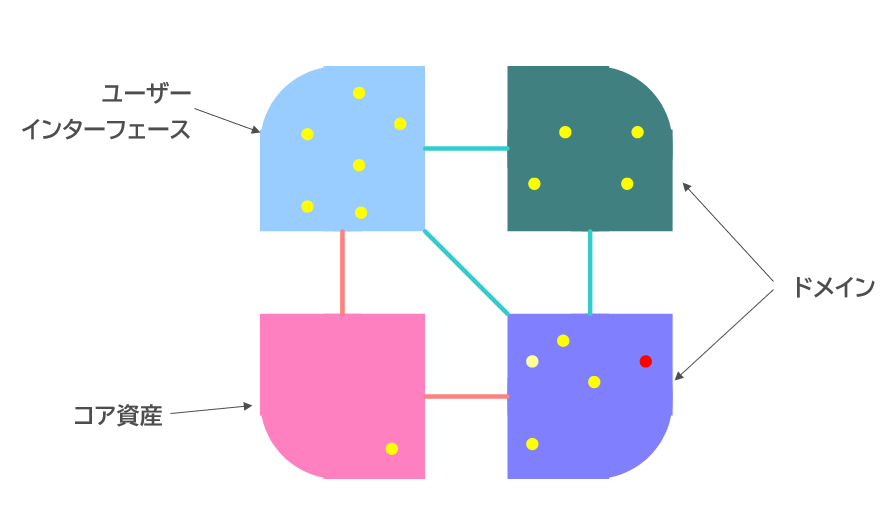
だから、ソフトウェアのアーキテクチャを設計する際に、リスクが高いモジュールとリスクが低いモジュールを分離しておかないといけない。リスクが高いモジュールとは、多くの場合、診断や治療を担うソフトウェアモジュールのこととなる。診断や治療を担うソフトウェアが期待通りに働かないと、誤診したり、意図しない出力を患者に与えたりするからだ。
また、例えば、意図しない出力を出していないかどうかを判定するリスクコントロール手段としてのソフトウェアや、セルフチェック機能が誤動作して、医療機器の機能が使えなくなるような場合のソフトウェアもソフトウェア安全クラスはBやCとなる。
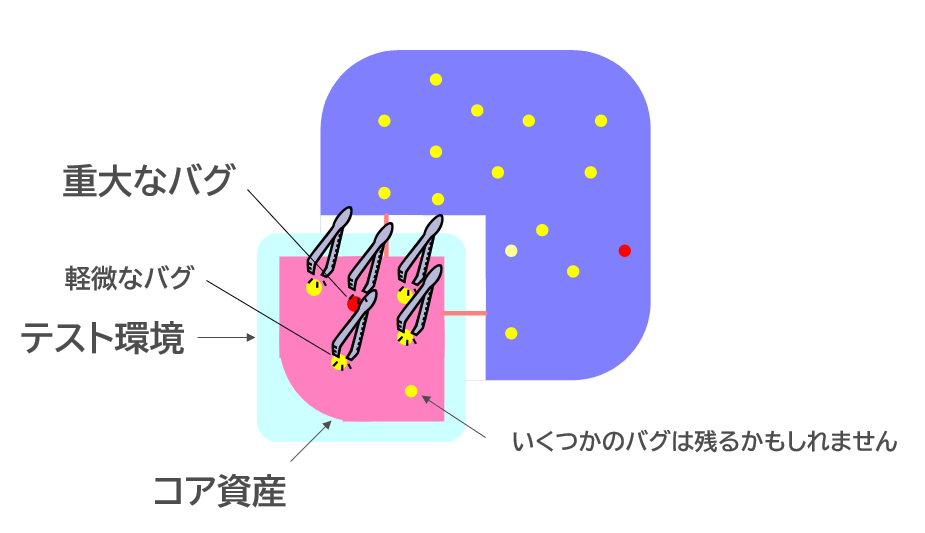
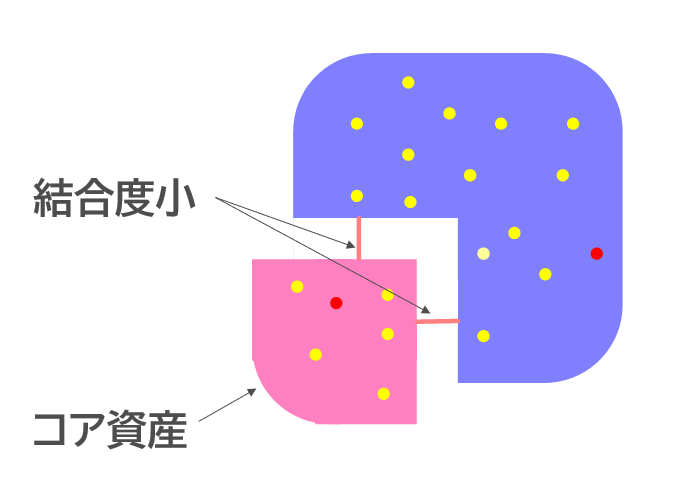
だから、医療機器ソフトウェアを開発するときは、フィージビリティスタディ(要素技術開発やコンセプト開発)のフェーズでも、リスクの高いモジュールは、リスクの低いモジュールと分離して、高凝集、疎結合にしておかないといけないのだが、医療機器できるかどうか、臨床的に効果が見込めるかどうか分からない段階では、まず、どんなものでもいいから、動くソフトウェアが欲しくて、医療機器ソフトウェアの開発経験が無い、IEC 62304 の規格要求も知らないソフトウェアハウスに、とりあえずソフトウェアを作ってもらってしまう。

実際、臨床効果が見込めなければ、資金調達も難しいだろうし、最初から完璧な規格適合ができるようなリソースも用意できない状況では、取りあえず動くソフトウェアを作るというアプローチを取るのも仕方がないだろう。
しかし、臨床的にも「これはいける」となって、資金も調達できる見込みが立ったときに、CEOはソフトウェアを作り直すという決断はしないだろう。おそらく、動いているんだから、試作レベルで作ったソフトウェアをそのまま製品にしろ、テストでバグが見つかったら修正すればよいと考えるはずだ。
でも、そんな考えでは、IEC 62304 の規格には適合できないし、そもそも、市場に出したときに患者危害に至るような障害が発生する危険性が高まる。本当に障害が発生すれば、リコールになって膨大な回収費用を被ることになる。1回も痛い目にあっていない経営者には、その痛みは分からないかもしれない。
リコールのコストもさることながら、患者危害に至れば、国民の安全を守ることはできないから、各国の規制当局は医療機器ソフトウェアの市販前申請の際に、キチンと規格に適合しているかどうかをチェックする。
ソフトウェアは中身が見えないから、チェックするレビューワーの技量にもよってしまうが、まともにアーキテクチャ図が描けていないような場合は、米国FDAなら、まず、申請は通らない。
じゃあ、どうすればよいかを解説したのが今回の動画になる。
ソフトウェアの詳細な技法のことを説明してもピンとこないと思ったので、紙芝居風にしてイメージを掴んでもらうことにした。
ざっとこんな感じだ。
一度作ってしまったソフトウェアシステムのイメージ



これは、今や医療機器は単独で動くのではなく、データをネットワーク経由で他のシステムに送信したり、クラウドに送ってAI解析したりすることが当たり前になって、どのソフトウェア機能が何のハードウェアや他のソフトウェアとつながって、診断、治療、予防を担っているのかを把握した上で審査しなければならなくなってきたからである。
下記布図は、FDAの医療機器ソフトウェア機能の市販前申請の内容ガイダンスの附属書に掲載されている 3つの仮想の医療機器のシステム&ソフトウェアアーキテクチャ図を UMLツール EnterpriseArchitectでトレースしなおした図だ。



- ハードウェアモジュール、ソフトウェアモジュール、OTS(市販ソフトウェア)、その他のモジュール(医療機器の基本機能ではないモジュール)の種別と、それらのモジュール間のインタフェースの方向性が知りたい。
- 各モジュールには、要求仕様があるはずなので、それぞれのモジュールに要求仕様のIDを付与してトレースできるようにする。
- 当該医療機器本体だけでなく、ネットワーク接続できる周辺機器やクラウドサービスを明示させる。
そんな動画を作っていたら、たまたま、Software Design 2025年8月号の特集記事が『第1特集 そのリファクタリング、今やるか?見送るか?適切なタイミングとビジネス面の価値』だった。こちらは IT業界のリファクタリングの話しらしい。
医療機器業界の方には、是非、医療機器ソフトウェアのリファクタリングの動画を見ていただきたい。(18分)
0 件のコメント:
コメントを投稿